Photon and neutron-based techniques for studying membrane dynamics and protein aggregation in lipid–protein interactions

1
Department of Raman Spectroscopy, Frank Laboratory of Neutron
Physics, Joint Institute for Nuclear Research, Dubna, Russia
2
Department of Biophysics, Faculty of Science, Cairo University,
Giza, Egypt
3
Academy of Scientific Research and Technology (ASRT), Cairo,
Egypt
Abstract
Lipid–protein interactions are central to maintaining the structural and functional balance of biological membranes, influencing a wide array of cellular processes. These interactions, however, become pathological in neurodegenerative diseases (NDDs), such as Alzheimer's, Parkinson's, and Huntington's diseases. In these disorders, the misfolding and aggregation of proteins like amyloid-beta (A\(\beta )\), alpha-synuclein (\(\alpha \)-syn), and mutant huntingtin (mHTT) disrupt the lipid bilayer, compromising membrane integrity, fluidity, and signaling. In this review we explore the critical role of lipid–protein interactions in NDDs, emphasizing how protein misfolding leads to toxic aggregates that embed into membranes, triggering neurotoxic events. Advanced spectroscopic techniques have been instrumental in studying these molecular interactions. Photon-based methods, including Förster resonance energy transfer (FRET), circular dichroism (CD), and Raman spectroscopy, provide real-time insights into protein aggregation and lipid membrane dynamics. Neutron-based techniques, such as neutron reflectometry and small-angle neutron scattering (SANS), further enhance the resolution of lipid–protein interactions, particularly in the context of neurodegenerative aggregation.
Moreover, the review highlights the significance of lipid microdomains, particularly cholesterol-rich lipid rafts, which act as platforms for protein aggregation, influencing disease progression. Therapeutic strategies aimed at targeting these lipid–protein interfaces are also discussed, with a focus on how spectroscopic insights have driven the development of drugs that stabilize membrane integrity or prevent toxic aggregation. Finally, the integration of spectroscopy with computational models, such as molecular dynamics (MD) simulations, is proposed as a promising approach to further unravel the complex dynamics of lipid–protein interactions, providing a more complete picture of disease mechanisms.
Keywords: lipid–protein interactions; neurodegenerative diseases; amyloid-β; protein aggregation; protein secondary structure, SERS, CARS, SANS, MD simulation
1. Introduction
Lipid–protein interactions are fundamental to the orchestration of cellular life, particularly within the dynamic architecture of biological membranes [1,2,3]. These interactions are not merely structural but function as complex regulatory mechanisms, influencing an array of processes essential to cellular integrity and communication [4,5]. The lipid bilayer — far from being a passive scaffold — influences protein function, stability [6], and localization. In return, membrane proteins modulate the physical properties of the bilayer [7], affecting membrane fluidity, curvature, and microdomain formation [8]. This molecular crosstalk is especially crucial in maintaining the delicate balance of neuronal function [9], where any disruption in lipid–protein interactions can have catastrophic consequences [10,11], contributing to neurodegeneration and the aggregation of toxic protein species [12].
The involvement of lipid–protein interactions in neurodegenerative diseases, such as Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD), has increasingly come into focus, shedding light on their critical role in disease pathology [13]. Neurodegeneration is often marked by protein misfolding and aggregation [14,15], where proteins such as A\(\beta \) [16], \(\alpha \)-syn [17,18], and mHTT [19] form toxic oligomers that disrupt cellular homeostasis. These protein aggregates not only disturb neuronal signaling but also severely compromise membrane integrity, leading to altered lipid–protein interactions. The lipid bilayer becomes a battleground for pathological changes as these misfolded proteins embed themselves into membranes, disturb ion homeostasis, and trigger downstream neurotoxic effects [20].
In Alzheimer's disease, the aggregation of A\(\beta\) peptides within lipid bilayers alters membrane fluidity and permeability [21], disrupting calcium signaling and causing synaptic dysfunction [22]. A\(\beta\) preferentially associates with lipid rafts — cholesterol and sphingolipid enriched microdomains — where it interferes with essential signaling cascades, exacerbating neuronal toxicity. In Parkinson's disease, \(\alpha \)-syn, a protein normally involved in synaptic vesicle trafficking, misfolds and aggregates at the membrane surface [23], disrupting membrane curvature and leading to synaptic failure. Huntington's disease follows a similar pattern, where mHTT aggregates sequester membrane-associated proteins [24], impairing intracellular trafficking and autophagy. Across these diseases, common themes emerge: protein aggregation, lipid disruption, and the collapse of membrane–protein interactions, fuelling the progression of neurodegeneration [25].
Understanding the molecular underpinnings of lipid–protein interactions and their pathological derailments in neurodegeneration demands advanced investigative tools. In this regard, spectroscopic techniques have emerged as invaluable in elucidating the structural and dynamic intricacies of these interactions [26,27]. Both photonic and neutron-based spectroscopic methods have provided researchers with powerful means to explore how proteins behave within lipid environments, how these environments influence protein misfolding and aggregation, and how neurodegenerative diseases alter these delicate systems [28].
Photon-based techniques have revolutionized our ability to study lipid–protein interactions with high spatial and temporal resolution [29]. Fluorescence spectroscopy is a widely used tool that exploits the sensitivity of fluorophores to monitor protein movements [30] conformational changes, and interactions with lipids in real time. Techniques such as FRET [31] fluorescence correlation spectroscopy enable the detection of subtle changes in protein–lipid associations, particularly within membrane microdomains like lipid rafts [32]. These methods are particularly useful in studying how disease-related proteins such as A\(\beta\) or \(\alpha\)-syn interact with membranes [33], highlighting the role of lipid composition modulating toxic protein aggregation.
In parallel, infrared (IR) spectroscopy, particularly attenuated total reflectance Fourier-transform infrared (ATR-FTIR) spectroscopy, provides insights into the secondary structure of proteins as they interact with lipid bilayers [34,35]. This technique detects molecular vibrations that reflect changes in protein conformation, making it a powerful tool for studying protein misfolding and aggregation. In the context of neurodegenerative diseases [36], IR spectroscopy has been instrumental in tracking the structural transitions of A\(\beta\) and \(\alpha\)-syn from their soluble forms into toxic oligomers and fibrils within membrane environments [37,38]. These structural insights are critical for understanding how the lipid environment contributes to or hinders the progression of protein aggregation.
CD spectroscopy further complements these methods by allowing researchers to probe the overall secondary structure of membrane proteins and their lipid-induced conformational changes [39]. CD spectroscopy is particularly useful for monitoring the folding behaviour of proteins like A\(\beta \) as they transition into pathological aggregates upon interaction with lipid membranes [40,41]. By measuring differential absorption of circularly polarized light, this technique provides valuable information about how proteins shift between \(\alpha \)-helical and \(\beta \)-sheet rich structures — transitions often associated with the formation of neurotoxic aggregates [42].
Neutron-based techniques, such as neutron reflectometry and SANS, offer unique advantages in studying lipid–protein interactions [43], particularly within membrane systems [44]. Neutron reflectometry allows for the precise measurement of lipid bilayer thickness and protein insertion depth, providing a detailed view of how proteins integrate into or perturb the membrane. This technique is highly sensitive to the contrast between different molecular components, making it ideal for investigating how neurodegenerative proteins like \(\alpha \)-syn or mHTT interact with lipid bilayers. Neutron scattering methods, such as SANS, further enhance our ability to study protein aggregation in lipid environments by offering insights into the size, shape, and distribution of protein aggregates within membrane systems. These neutron-based approaches are especially powerful when combined with deuterium labelling, allowing researchers to distinguish between lipid and protein components with high resolution.
Raman spectroscopy as another valuable, strategic photon-based technique has found increasing application in the study of lipid–protein interactions [45]. Raman spectroscopy detects molecular vibrations, much like IR spectroscopy, but with the added advantage of being non-destructive and providing high spatial resolution [46]. This method has been applied to investigate the interaction of misfolded proteins with lipid membranes, helping to elucidate the molecular basis of neurodegenerative diseases at the membrane interface.
In this review, we will explore how these advanced spectroscopic techniques — spanning both photonic and neutron-based methods — have shed light on the molecular choreography of lipid–protein interactions and their derailment in neurodegenerative diseases [47]. By uncovering the intricate details of how lipids and proteins communicate within the membrane, these methods not only provide a deeper understanding of the fundamental biology of membranes but also offer promising avenues for therapeutic intervention. The ability to visualize and manipulate lipid–protein interactions at such a fine molecular level offers new hope for the development of strategies aimed at preventing or reversing the toxic aggregation of proteins that drive the progression of neurodegeneration. Through the lens of spectroscopy, we are poised to unlock the mysteries of membrane biology and its implications for human health, offering a path forward in the fight against devastating neurodegenerative disorders.
2. Photon-based spectroscopic techniques for investigating protein–lipid interactions
Photon-based spectroscopic methods involve the use of light to study the properties of molecules (Figure 1). When photons interact with a sample, they can be absorbed, scattered, or emitted, providing information about the molecular structure, dynamics, and environment [48]. In the context of membrane–protein interactions, photon-based techniques allow researchers to
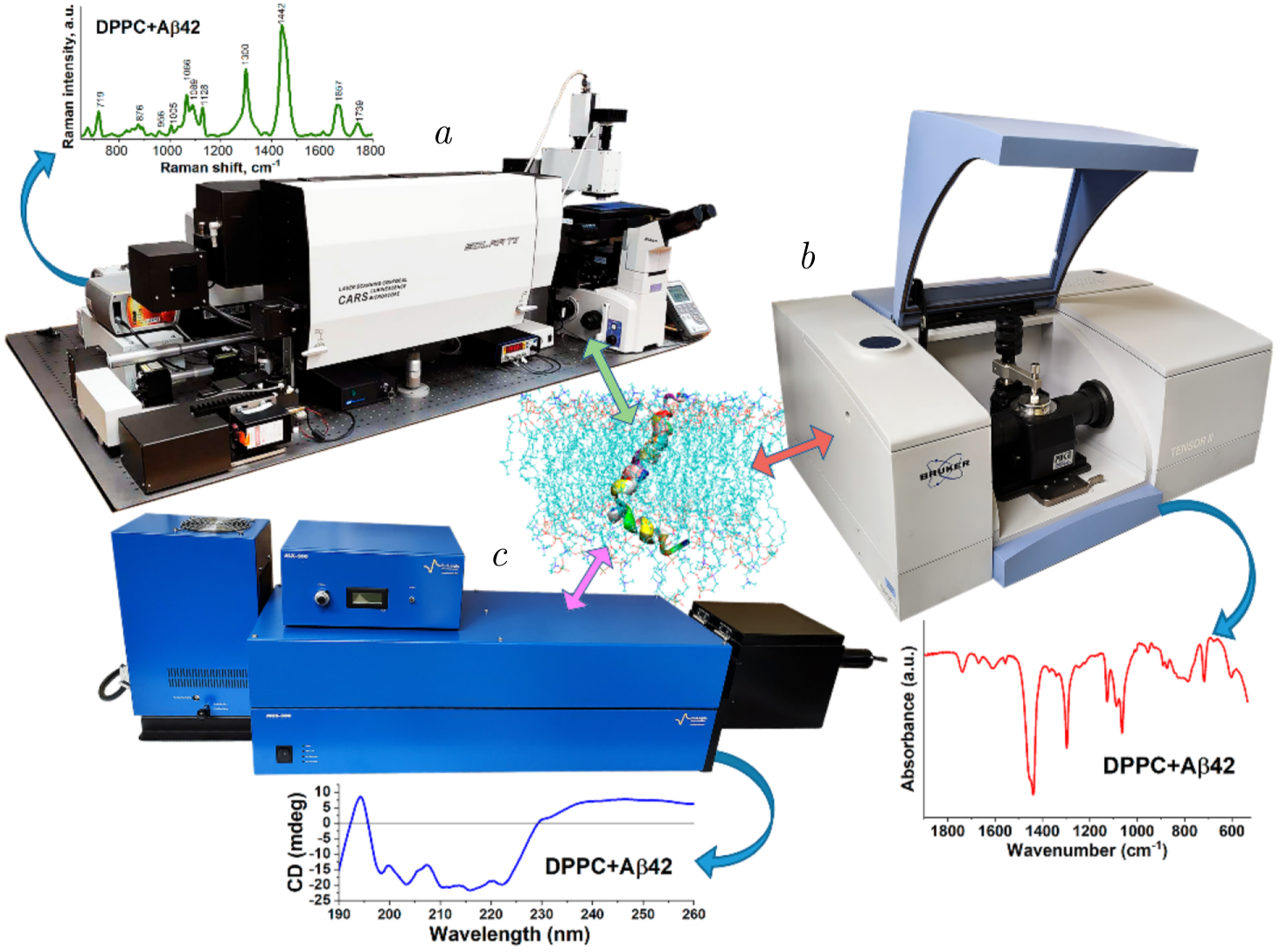
Figure 1. The illustration of the photon-based spectroscopic facilities utilized at the Sector of Raman Spectroscopy, FLNP, JINR, for advancing lipid–protein interaction studies. The Confotec CARS Raman microspectrometer (SOL Instruments, Belarus) (\(a\)) enables high-resolution molecular imaging and vibrational analysis, providing chemical and structural insights into lipid and protein assemblies. The TENSOR II FTIR spectrometer (Bruker, Germany) (\(b\)) facilitates the examination of secondary structure changes in proteins and lipid organization through infrared absorption, revealing interaction dynamics. The MOS-500 circular dichroism spectrometer (BioLogic, France) (\(c\)) is employed to assess protein folding and conformational changes under the influence of lipid environments. Together, these instruments allow a comprehensive investigation of lipid–protein interactions at molecular and structural levels.
observe changes in the lipid bilayer or protein conformation in real time, often without the need for invasive sample preparation. These techniques are especially valuable for studying systems at physiological conditions [49].
Photon scattering refers to the redirection of light as it encounters molecules. The scattered light can reveal critical information about the structure and dynamics of the scattering particles [50,51]. In membrane–protein interaction studies, two forms of photon scattering are particularly [52]: Rayleigh scattering and Raman scattering. For Rayleigh scattering, it is being defined as the elastic scattering of light, where the scattered photons have the same energy as the incident photons. Raman scattering, on the other hand, is an inelastic scattering process where the scattered photons have an energy different from that of the incident photons. This energy difference is due to the interaction of the incident light with molecular vibrations, causing the photons to gain or lose energy. This technique can provide insights into the size, shape, and organization of membrane-associated proteins or protein–lipid complexes [53].
2.1. Raman spectroscopy mechanism and advantages in studying lipid–protein interactions
Raman spectroscopy is a highly valuable tool in the study of lipid–protein interactions, offering detailed insights into the molecular vibrations of these biological molecules (Figure 2). By detecting inelastic scattering of photons, it provides information about the vibrational states of molecular bonds, which is useful for probing the conformational changes in proteins and lipids [54]. In the context of studying membrane–protein interactions, Raman spectroscopy is particularly advantageous because it requires minimal sample preparation, can be applied to complex biological systems, and allows for non-destructive analysis [55].
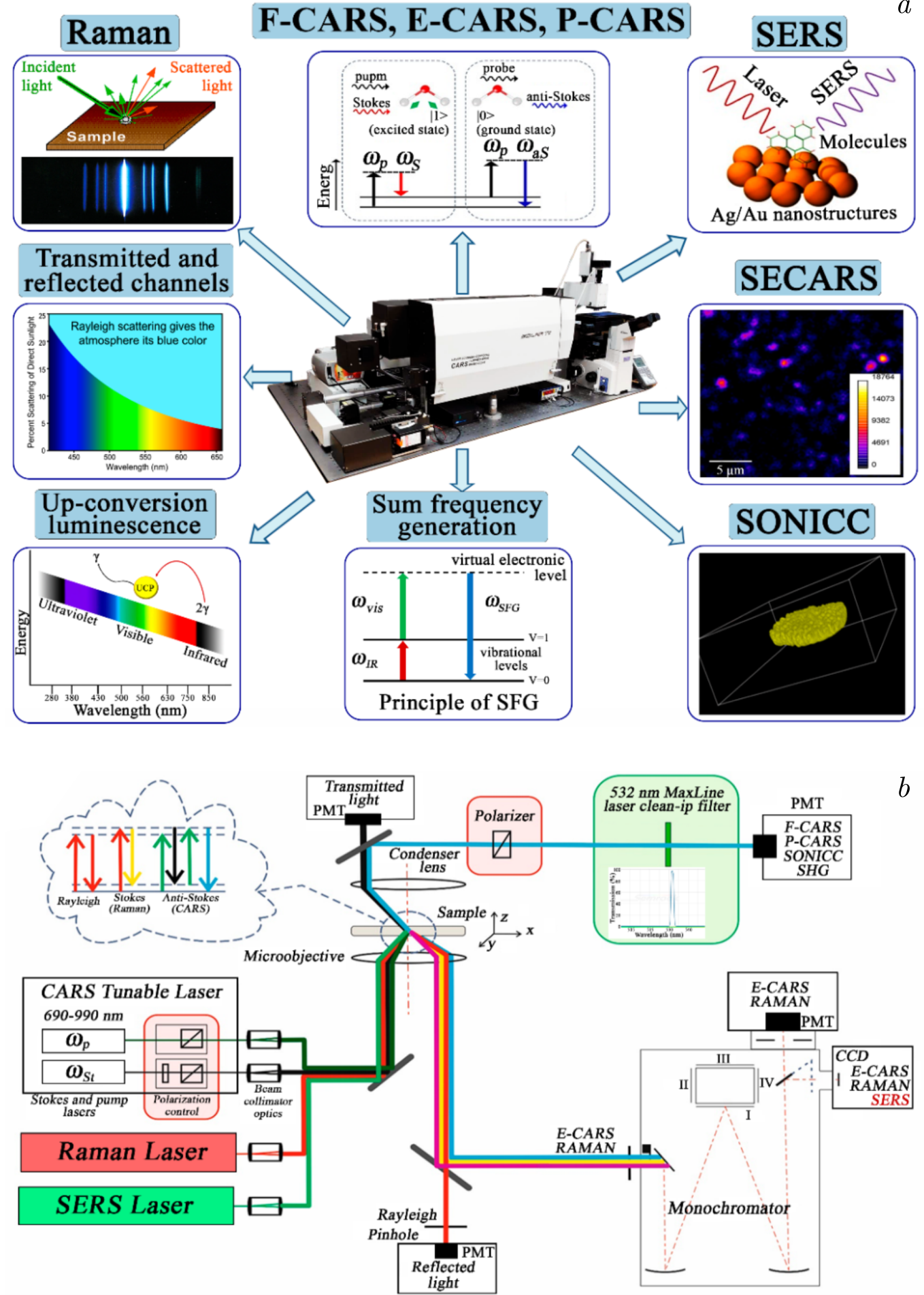
Figure 2. a) Multimodal optical platform based on the Confotec CARS Raman microspectrometer, and b) optical layout used for lipid–protein interaction studies, where spontaneous Raman spectroscopy is employed alongside with CARS (Coherent anti-Stokes Raman spectroscopy) [58] and SERS (Surface-Enhanced Raman Spectroscopy) for enhanced analysis [59].
The underlying mechanism of Raman spectroscopy revolves around the scattering of light. When light encounters a molecule, most photons are elastically scattered, known as Rayleigh scattering. However, a small portion of the light undergoes inelastic scattering, resulting in a change in the energy of the photons — this is Raman scattering [56]. The energy shift corresponds to the vibrational modes of the molecular bonds within the sample, providing a "fingerprint" that reflects the chemical structure of molecules like proteins and lipids. These vibrational modes, such as C–H, N–H, and C\(=\)C stretches, are directly linked to the molecular conformation and dynamics of these biological molecules. This ability to probe molecular vibrations without altering the natural state of the sample makes Raman spectroscopy particularly advantageous in studying lipid–protein interactions within intact membranes [57].
The high chemical specificity of Raman spectroscopy allows for precise differentiation between various lipid species and protein conformations. By analysing distinct vibrational signatures, researchers can distinguish between different lipid environments and monitor conformational changes in proteins. This is particularly beneficial in studying membrane–protein interactions, where subtle alterations in lipid composition or protein structure can have profound biological consequences. Moreover, because Raman spectroscopy does not require the use of external labels or probes, it preserves the native behaviour of proteins and lipids, providing a more accurate representation of biological systems in their natural states.
Raman spectroscopy is also highly compatible with biological membranes, enabling the study of lipid bilayers, membrane proteins, and lipid–protein interactions in near-physiological conditions. This makes it a valuable tool for exploring complex systems, such as lipid rafts or membrane-mimicking systems like liposomes. Its ability to operate in aqueous environments further enhances its suitability for studying biomolecules in their hydrated, functional states. The technique is particularly effective for probing the dynamics of both lipids and proteins, allowing researchers to investigate how protein activity influences lipid bilayer structure, as well as how lipid composition modulates protein conformation and function.
2.1.1. Raman spectroscopy case study in detecting lipid-induced A\(\beta \) misfolding in Alzheimer’s disease
One of the most significant applications of Raman spectroscopy in lipid–protein research is its use in studying the interactions between A\(\beta \) peptides and lipid membranes in Alzheimer's disease. A\(\beta \) aggregation is a hallmark of Alzheimer's pathology, and lipids are known to play a critical role in modulating the misfolding and aggregation of A\(\beta \) peptides [60]. The interaction of A\(\beta \) with lipid membranes triggers the misfolding of these peptides into \(\beta \)-sheet-rich structures, which form toxic aggregates that contribute to neuronal damage and synaptic dysfunction. By monitoring changes in the amide bands of A\(\beta \) peptides, Raman spectroscopy can detect these conformational transitions and provide valuable insights into the mechanisms underlying A\(\beta \) aggregation [61].
Specifically, Raman spectroscopy has revealed that certain lipid environments, particularly those rich in cholesterol and sphingolipids, promote the formation of \(\beta \)-sheet structures in A\(\beta \) [62]. This finding is critical because the \(\beta \)-sheet conformation is associated with the formation of toxic fibrils that exacerbate Alzheimer's disease [63]. Raman spectra capture the distinct vibrational frequencies corresponding to different conformations of A\(\beta \), allowing researchers to track the progression of peptide misfolding and aggregation (Figure 3) [61]. Additionally, lipid composition plays a crucial role in this process — cholesterol-containing membranes tend to accelerate A\(\beta \) aggregation, while unsaturated lipids inhibit it. Raman spectroscopy's ability to simultaneously probe lipid and protein dynamics offers a unique perspective on how lipid composition influences A\(\beta \) behaviour, contributing to our understanding of Alzheimer's disease pathogenesis [64].
Beyond the aggregation of A\(\beta \), Raman spectroscopy can also detect lipid peroxidation, a common feature of neurodegenerative diseases like Alzheimer's. Oxidative stress leads to the degradation of lipid membranes, further exacerbating A\(\beta \) aggregation and creating a destructive
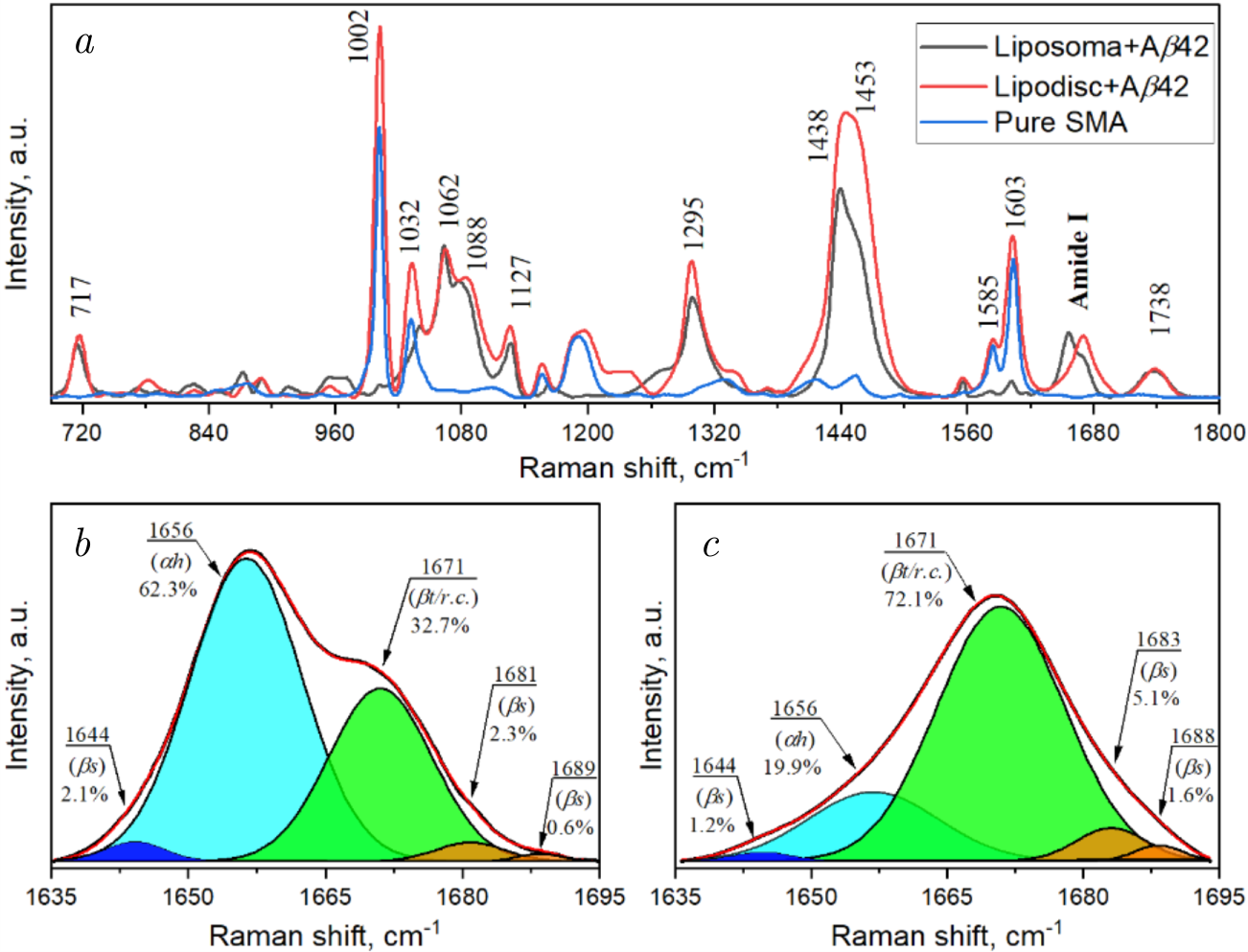
Figure 3. Raman spectra exemplifying protein–lipid interactions involving amyloid beta and 1,2-dimyristoyl-sn-glycero-3-phosphocholin (DMPC) membranes: (\(a\)) Raman spectra of styrene-maleic acid (SMA) (blue line), amyloid beta with DMPC liposomes (black line), and amyloid beta with DMPC lipodiscs (red line) at a molar ratio (SMA/DMPC) of 1.28. Deconvolved Raman spectra of amyloid beta in the Amide I region: (\(b\)) in DMPC liposomes, (\(c\)) in DMPC lipodiscs. The red line represents the fitted curve, while the filled areas correspond to deconvolved spectra using the Gaussian function. \(\alpha h\) — \(\alpha \)-helix, \(\beta t/r.c\).— \(\beta \)-turn/random coil, \(\beta s\) — \(\beta \)-strand. The figure is reprinted with the kind permission of the publisher, license number 5935770613802.
feedback loop. By monitoring the vibrational modes associated with lipid peroxidation, Raman spectroscopy provides a direct link between oxidative damage and A\(\beta \) aggregation, offering new avenues for studying the molecular mechanisms of neurotoxicity [65].
To enhance the sensitivity and resolution of Raman spectroscopy in studying membrane–protein interactions, advanced techniques such as Raman imaging and SERS have been developed. Raman imaging combines spectroscopy with microscopy, enabling spatially resolved measurements of lipid–protein interactions at the cellular level. This technique allows researchers to map the distribution of A\(\beta \) aggregates within lipid-rich regions of cells, providing valuable insights into the localization and behaviour of these toxic species within neuronal membranes. By visualizing the interaction of A\(\beta \) with lipid domains, Raman imaging offers a powerful tool for studying the early stages of AD [66].
SERS, on the other hand, significantly enhances the Raman signal by using metallic nanoparticles or roughened metal surfaces. This enhancement allows for the detection of extremely low concentrations of biomolecules, making it ideal for studying weak lipid–protein interactions or early-stage A\(\beta \) aggregation [67]. SERS has been successfully applied to detect toxic oligomeric species of A\(\beta \), which are difficult to observe with conventional techniques [68]. This capability makes SERS a valuable tool for studying the initial stages of protein misfolding and aggregation, offering potential for early diagnosis and intervention in neurodegenerative diseases [69].
Therefore, Raman spectroscopy and its advanced variants provide a powerful and versatile toolkit for studying lipid–protein interactions. Its ability to offer detailed molecular insights in a non-invasive and label-free manner, combined with its compatibility with complex biological systems, positions it as a crucial technique for exploring the molecular underpinnings of diseases like Alzheimer's. The expanding applications of Raman imaging and SERS further enhance its potential, enabling researchers to explore new dimensions of lipidcprotein dynamics with unprecedented sensitivity and resolution [70].
2.2. Fluorescence spectroscopy
Fluorescence spectroscopy is an essential tool in the study of membranevprotein interactions, widely recognized for its sensitivity, real-time monitoring capabilities, and non-invasive nature [71]. The method relies on the detection of emitted light from fluorophores that have absorbed energy at specific wavelengths. In the context of membrane–protein systems, fluorescence spectroscopy provides detailed information about the spatial and temporal dynamics of proteins and lipids, revealing key insights into protein conformations, membrane dynamics, and the proximity of proteins to lipid bilayers [72].
Among the most widely applied fluorescence techniques for studying membrane–protein interactions is Förster Resonance Energy Transfer (FRET) [73], which is particularly useful for monitoring the proximity of molecules on a nanometre scale. FRET occurs when energy is transferred from a donor fluorophore to an acceptor fluorophore in close proximity, typically within a range of 1–10 nm [74]. This energy transfer is highly sensitive to distance, making FRET an ideal tool for detecting nanoscale changes in molecular arrangements and interactions. In the context of membrane–protein studies, FRET can illuminate critical aspects such as how proteins associate with lipid bilayers, undergo conformational changes upon binding, and engage in protein–protein interactions at or near membranes [75].
FRET has proven especially valuable for studying membrane association. By labelling a soluble protein with a donor fluorophore and the lipid membrane with an acceptor fluorophore, the technique can detect whether a protein has inserted into or is merely associated with the membrane [71]. An increase in FRET signal suggests close contact or membrane insertion, while a decrease in signal may indicate separation between the protein and the membrane. This allows researchers to track dynamic membrane association events in real time, contributing to a more comprehensive understanding of membrane-bound processes.
Furthermore, FRET is an excellent method for investigating protein conformational changes, particularly those induced by interactions with lipid membranes. By labelling two sites within a protein, FRET can monitor structural rearrangements that occur as the protein binds to the membrane [76], oligomerizes, or undergoes folding/unfolding events [77,78]. The changes in FRET efficiency between donor and acceptor fluorophores reflect shifts in molecular distances, providing a powerful means of observing how membrane environments affect protein structure and function [79].
Beyond protein conformation, FRET also provides valuable insights into protein–protein interactions, especially for studying oligomerization or clustering on membrane surfaces [80,81]. Membrane-bound proteins often form complexes essential for processes such as signal transduction, and FRET allows researchers to measure the proximity between labelled proteins to determine whether they are assembling into functional clusters [82]. By detecting changes in FRET efficiency, it is possible to observe the dynamics of protein interactions in near-physiological conditions, advancing our understanding of membrane-associated signalling and other crucial cellular processes [83,84].
2.2.1. Fluorescence spectroscopy case study of alpha-synuclein interactions with lipid membranes in Parkinson’s disease
A prominent case where fluorescence spectroscopy, particularly FRET, has played a pivotal role is in the study of \(\alpha \)-syn interactions with lipid membranes, particularly in the context of Parkinson's disease. Alpha-synuclein is a small, intrinsically disordered protein that is essential for normal synaptic function but is also implicated in the pathogenesis of Parkinson's disease. In this neurodegenerative disorder, \(\alpha \)-syn aggregates into insoluble fibrils, forming toxic structures known as Lewy bodies. These aggregates are thought to contribute to neuronal death and synaptic dysfunction, and the protein's interactions with lipid membranes are believed to play a key role in its misfolding and aggregation [85,86].
FRET has been extensively employed to study how \(\alpha \)-syn interacts with lipid membranes under both physiological and pathological conditions [87,88]. Research using FRET has demonstrated that \(\alpha \)-syn adopts different conformations depending on the curvature and composition of the lipid bilayer. For example, when \(\alpha \)-syn binds to highly curved membranes, such as those found in synaptic vesicles, it assumes an extended helical conformation [89]. By labelling \(\alpha \)-syn with a donor fluorophore and the membrane lipids with an acceptor, FRET measurements can provide a detailed picture of how \(\alpha \)-syn interacts with the membrane surface, and how these interactions influence its conformation and aggregation.
Lipid composition also plays a crucial role in \(\alpha \)-syn aggregation, and FRET has provided valuable insights into how different lipid environments modulate this process. Studies using FRET have shown that certain lipid species, such as phosphatidylserine (PS) and gangliosides, promote \(\alpha \)-syn aggregation into toxic fibrils, whereas other lipids, like phosphatidylcholine (PC), tend to inhibit this process [90]. FRET experiments that label \(\alpha \)-syn and membrane lipids reveal that specific lipid compositions facilitate the formation of membrane-bound oligomeric species of \(\alpha \)-syn, which are more toxic than the fibrillar aggregates. These oligomeric forms are thought to disrupt membrane integrity by forming pores, leading to ion dysregulation and cellular toxicity, a key feature of Parkinson's disease pathology [91].
In addition to monitoring \(\alpha \)-syn's interaction with lipids, FRET has been used to investigate the oligomerization of \(\alpha \)-syn on the membrane surface, which is a crucial step in its toxic aggregation. By labelling different \(\alpha \)-syn molecules with donor and acceptor fluorophores, FRET can measure the proximity between them, providing evidence of oligomer formation. These early-stage oligomers, detected by an increase in FRET efficiency, are believed to be particularly toxic, capable of permeabilizing membranes and inducing cellular dysfunction [92]. Moreover, the curvature of the lipid bilayer has been shown to influence the extent of \(\alpha \)-syn oligomerization, with small, highly curved vesicles inducing more significant oligomerization than larger or flatter membranes. This suggests that \(\alpha \)-syn may preferentially aggregate in regions of high membrane curvature, such as synaptic vesicles, which could explain its pathogenic effects in neurons.
Another fluorescence technique, Fluorescence Recovery After Photobleaching (FRAP), complements FRET by offering additional insights into the mobility of membrane-bound \(\alpha \)-syn. FRAP measures the lateral mobility of fluorescently labelled \(\alpha \)-syn on membranes by bleaching a specific region and monitoring the recovery of fluorescence [93]. This technique has demonstrated that membrane-bound \(\alpha \)-syn forms stable oligomeric structures that are significantly less mobile than their monomeric counterparts, further implicating membrane interactions in \(\alpha \)-syn aggregation and toxicity.
Therefore, fluorescence spectroscopy, particularly FRET, provides a powerful and versatile approach for exploring membrane–protein interactions in both physiological and pathological contexts. The detailed information obtained from fluorescence techniques regarding the proximity, conformational changes, and oligomerization of proteins like \(\alpha \)-synuclein in relation to lipid membranes has significantly advanced our understanding of neurodegenerative processes. As our knowledge of fluorescence methods continues to expand, these techniques will remain at the forefront of research into protein–membrane interactions, offering new insights into disease mechanisms and potential therapeutic targets.
2.3. Circular dichroism spectroscopy
Circular Dichroism (CD) spectroscopy is a powerful and widely used technique for analyzing the structural properties of chiral molecules, particularly in the study of proteins, nucleic acids, and other biomolecules. Chiral molecules, which are non-superimposable on their mirror images, interact differently with left- and right-circularly polarized light [94]. CD spectroscopy measures the difference in the absorption of these two types of polarized light as it passes through a sample. This differential absorption is expressed as a function of wavelength, thereby providing valuable insight into the structural aspects of the molecule, including its secondary, tertiary, and quaternary configurations [95].
One of the primary applications of CD spectroscopy is in the analysis of protein secondary structures, where it helps to distinguish between alpha-helices, beta-sheets, and random coils [96,97]. The far-UV region of the CD spectrum (190–250 nm) is particularly sensitive to the peptide bond environment, making it ideal for probing the secondary structure of proteins [98]. For example, alpha-helical structures exhibit characteristic negative bands near 208 and 222 nm, while beta-sheets show a negative band near 218 nm and a positive band near 195 nm (Figure 4). Random coils, on the other hand, display a relatively featureless spectrum with a weak negative band around 195 nm [99,100]. By comparing experimental spectra to
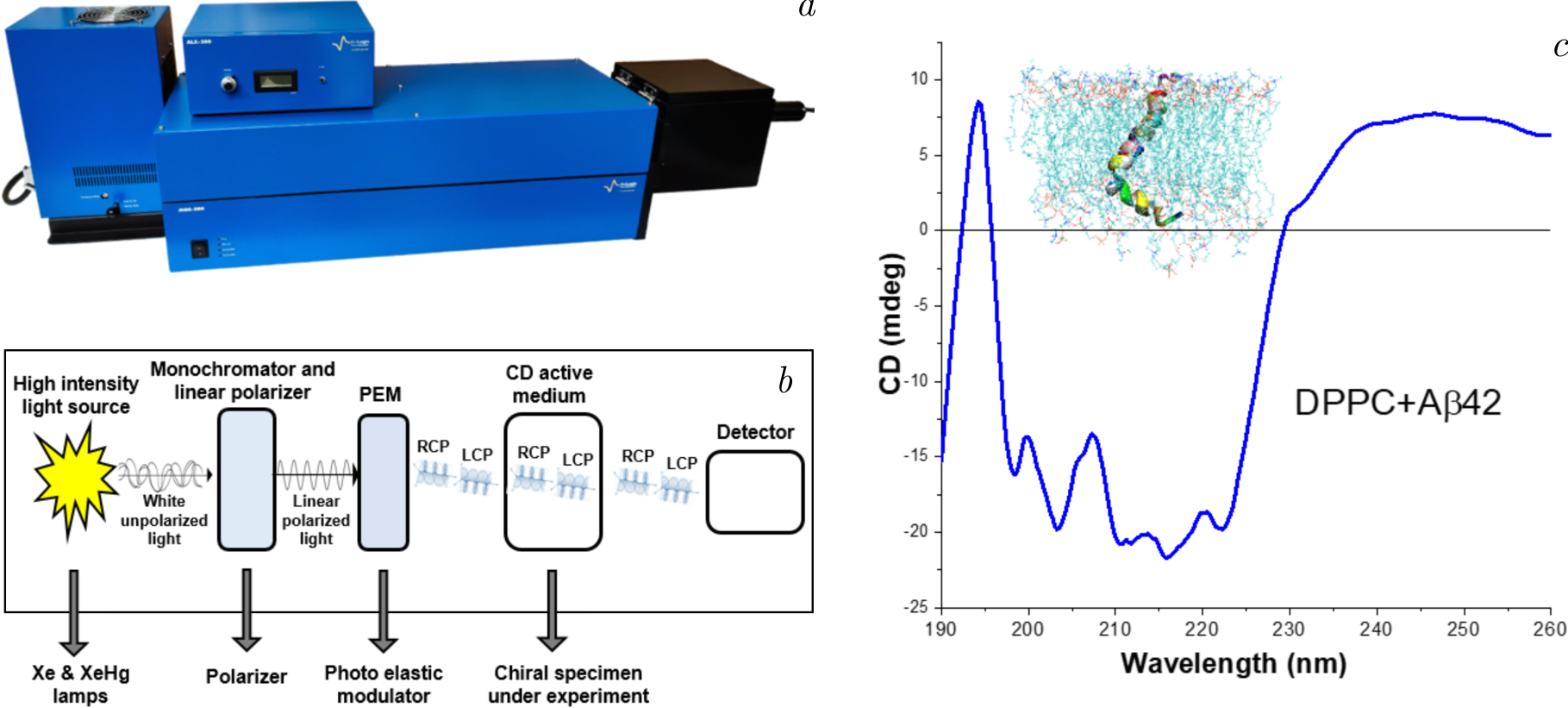
Figure 4. The illustration depicts the integration of circular dichroism spectroscopy into investigations of lipid–protein interactions. Panel (\(a\)) displays the MOS-500 CD spectrometer, an advanced tool designed to detect structural and conformational changes in proteins. Panel (\(b\)) illustrates the optical layout of the instrument, emphasizing the pathway of polarized light through the sample for precise measurements of chirality and secondary structure. Panel (\(c\)) showcases CD spectra of the Aβ42 peptide within a 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) lipid bilayer system, highlighting conformational shifts induced by the lipid environment. This figure underscores the utility of CD spectroscopy in elucidating peptide–lipid interactions critical to understanding biomolecular dynamics.
reference spectra of known structures, CD spectroscopy enables the estimation of the relative proportions of these structural elements in a protein, thus providing a detailed picture of its conformational state.
CD spectroscopy is also useful in monitoring protein folding and unfolding processes. Changes in the CD spectrum over time, as a protein is subjected to denaturing conditions such as heat or chemical agents, can reveal how the secondary structure evolves during folding or unfolding [101]. This is crucial for understanding the thermodynamics and kinetics of protein folding, as well as for studying diseases associated with protein misfolding, such as Alzheimer's and Parkinson's diseases. Additionally, CD spectroscopy has proven to be valuable in the analysis of protein–ligand interactions. When a ligand binds to a protein, it can induce conformational changes that are detectable by shifts in the CD spectrum, particularly in the far-UV or near-UV regions (250–300 nm), where the side chains of aromatic amino acids and disulfide bonds contribute to the signal [102,103].
Beyond proteins, CD spectroscopy is also employed to study nucleic acids. DNA and RNA are inherently chiral molecules, and their CD spectra are influenced by their helical conformations [104]. In the far-UV region, the CD signals can be used to assess changes in the secondary structure of nucleic acids, such as the transition from a B-form to a Z-form in DNA, or the formation of complex tertiary structures like G-quadruplexes in RNA. The ability of CD spectroscopy to track structural changes makes it a valuable tool in drug development, where researchers aim to design molecules that can specifically bind to nucleic acids and modulate their function [105].
A key advantage of CD spectroscopy is its ability to provide structural information about biomolecules in solution, under near-physiological conditions [106]. This contrasts with techniques such as X-ray crystallography, which require the biomolecule to be in a crystalline form, and nuclear magnetic resonance (NMR) spectroscopy, which often requires high concentrations of the sample. CD spectroscopy is relatively fast, requires only small amounts of material, and can be applied to a wide range of samples, from small organic compounds to large macromolecular complexes [107,108]. However, one limitation of CD spectroscopy is its relatively low resolution compared to other structural techniques, as it provides an overall picture of the secondary structure but does not reveal atomic-level details. Despite this, CD spectroscopy remains a fundamental tool in biophysical chemistry and structural biology [109].
Technological advances have further expanded the capabilities of CD spectroscopy. For example, synchrotron radiation circular dichroism (SRCD) spectroscopy offers increased sensitivity and spectral range, allowing for more detailed analyses of protein secondary structure, especially at wavelengths below 190 nm, which are inaccessible with conventional light sources. SRCD can provide information on subtle structural features, such as the presence of extended beta strands or irregular turns, which are important for understanding complex folding mechanisms [110,111].
Correspondingly, circular dichroism spectroscopy is a versatile and essential technique in structural biology, with applications ranging from protein structure analysis to the investigation of folding dynamics and biomolecular interactions. Its ability to provide rapid, solution-based structural insights makes it indispensable in both fundamental research and drug discovery. Despite certain limitations in resolution, CD spectroscopy continues to evolve, with innovations such as SRCD enhancing its power to probe molecular structures in greater detail [112].
2.3.1. Case study of lipid–protein interactions analyzed by circular dichroism spectroscopy
In the study conducted by Kurakin et al. [115], in which circular dichroism had been utilized to analyze the secondary structure of A\(\beta \)(25–35) [113] within both bicelle-like structures (BLSs) and small unilamellar vesicles (SUVs), CD spectra were collected from a DMPC (0.5 wt%) \(+\) A\(\beta \)(25–35) sample, recorded at 10\(^{\circ}\)C (identified as BLSs) and 40\(^{\circ}\)C (identified as SUVs) across three heating-cooling cycles. To ensure accuracy, the spectra were corrected by subtracting the CD signal of extruded DMPC (0.5 wt%) unilamellar vesicles, filtered through a 500 A pore [114]. The resulting spectra revealed a pronounced minimum at 196 nm with a moderate depression in the 210–230 nm range, signifying a predominantly random coil structure.
Additionally, the spectra showed minor changes in mean residue ellipticity \([\theta ]\) at 196 nm and 215 nm, alongside a slight shift in the \([\theta ]\) minimum towards higher wavelengths at elevated temperatures (from 195 nm at 10\(^{\circ}\)C to 196.5 nm at 40\(^{\circ}\)C). The stability of the A\(\beta \)(25–35) secondary structure was further confirmed by the reproducibility of the spectra across three cycles. No significant spectral changes were observed when the temperature fluctuated between 10\(^{\circ}\)C and 40\(^{\circ}\)C.
The data indicated that at 10\(^{\circ}\)C, the BLS sample consisted of approximately 2–5% \(\alpha \)-helices, 15–38% \(\beta \)-sheets, 10–29% \(\beta \)-turns, and 39–68% random coils. In the SUV sample at 40\(^{\circ}\)C, the secondary structure ratios changed slightly, with \(\alpha \)-helices decreasing to 2–6%, while \(\beta \)-sheets, \(\beta \)-turns, and random coils increased to 25–39%, 12–25%, and 37–61%, respectively. These findings highlight that the majority of peptides adopt unordered secondary structures (random coils and \(\beta \)-turns) in both lipid environments [115].
2.4. Fourier-transform infrared spectroscopy
Fourier-Transform InfraRed (FTIR) spectroscopy is a powerful analytical technique used to obtain the infrared spectrum of absorption or emission of a solid, liquid, or gas [116]. This method plays a significant role in molecular spectroscopy, offering detailed information about the molecular composition, structure, and interactions of compounds. FTIR spectroscopy measures how different chemical bonds within a molecule absorb infrared radiation at various frequencies, providing a molecular "fingerprint" that is unique to each substance. Because of its versatility, FTIR is widely used in fields ranging from chemistry and materials science to biology and medicine, with applications in the analysis of organic and inorganic compounds, proteins, lipids, nucleic acids, and more [117].
The basic principle of FTIR spectroscopy lies in the interaction between infrared light and molecular vibrations (Figure 5). Molecules absorb infrared light at specific frequencies, corresponding to the vibrational energy levels of their chemical bonds. When a molecule absorbs infrared radiation, its chemical bonds vibrate more vigorously, resulting in stretching, bending, or twisting motions that are specific to the functional groups within the molecule. These molecular vibrations are detected as absorption peaks in an FTIR spectrum, where each peak represents a particular bond or group of atoms. The frequencies of these peaks, typically measured in wavenumbers (cm\(^{-1}\)), can be correlated with specific molecular structures, allowing scientists to identify the chemical composition of a sample [118].
One of the key advantages of FTIR spectroscopy is its ability to analyze a wide range of samples in various states — solids, liquids, and gases — without requiring extensive sample preparation. The technique is nondestructive and can be applied to complex mixtures, making it ideal for real-world applications. For instance, in organic chemistry, FTIR is used to identify functional groups such as alcohols, amines, carboxyl groups, and carbonyl compounds, based on their characteristic absorption frequencies. By comparing an unknown sample's FTIR spectrum to reference spectra, researchers can determine the molecular structure or verify the identity of a compound.
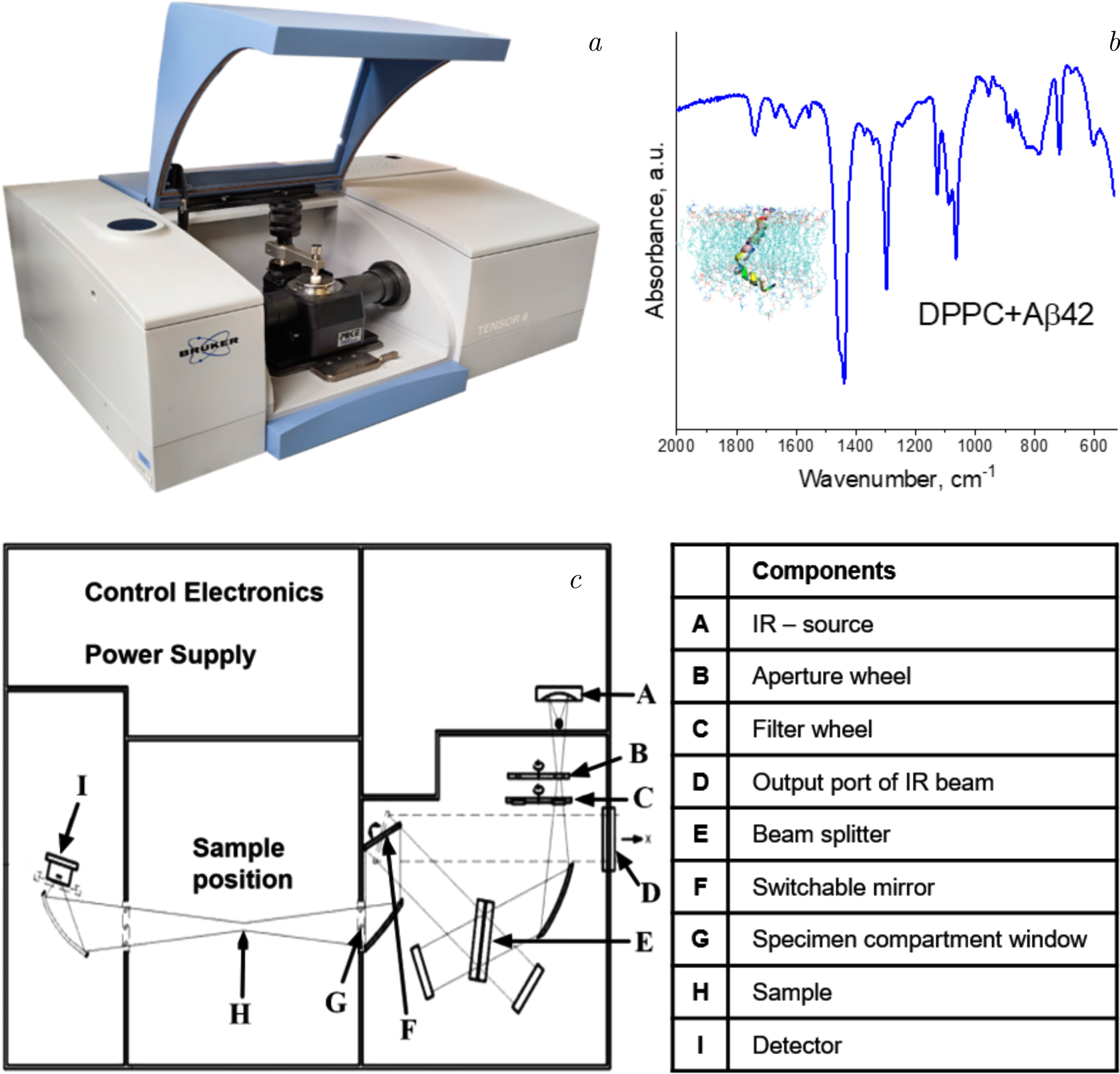
Figure 5. The illustration depicts the application of Fourier-transform infrared (FTIR) spectroscopy in the domain of lipid–protein interaction studies. Panel (\(a\)) shows the FTIR spectrometer setup, a powerful tool for investigating molecular vibrations and providing detailed information about the structural composition of lipid–protein systems. Panel (\(b\)) presents a representative FTIR spectrum of a lipid–protein system, demonstrating characteristic absorbance peaks that correlate with specific molecular vibrations of lipids and proteins, which can be used to assess structural changes and interactions. Panel (\(c\)) depicts the optical layout of the FTIR system, with a description of the beam path and sample configuration, highlighting the precision and sensitivity required to analyze the intricate interactions between lipid and protein components.
In biochemistry, FTIR spectroscopy is invaluable for studying biological macromolecules, particularly proteins, nucleic acids, and lipids [119]. For example, FTIR is frequently used to investigate protein secondary structures by analyzing the amide I and amide II bands in the infrared spectrum. The amide I band, which arises from the C\(=\)O stretching vibration of the protein backbone, is sensitive to the protein's secondary structure and can distinguish between \(\alpha \)-helices, \(\beta \)-sheets, and random coils. This allows researchers to monitor protein folding, conformational changes, or interactions with other biomolecules, such as ligands or membranes [120,121]. In a similar fashion, FTIR spectroscopy can be employed to study lipid structures and their phase behavior in membranes, providing insights into the dynamics of lipid–protein interactions [122].
FTIR is also extensively used in materials science to characterize polymers, ceramics, and other complex materials. For instance, in polymer science, FTIR can determine the degree of polymerization, the presence of cross-linking, or the incorporation of specific additives. Moreover, FTIR imaging and microscopy techniques have been developed, which allow spatially resolved chemical analysis of materials at the microscopic level. This is particularly useful in studying heterogeneous samples, where different regions of the material may have distinct chemical compositions [123].
A significant advancement in FTIR spectroscopy is the introduction of attenuated total reflectance (ATR) technology, which simplifies sample preparation and enhances the ease of obtaining spectra from solid or liquid samples. ATR-FTIR allows for the direct measurement of a sample without the need for complex transmission setups, making it highly accessible for routine analyses. This method is particularly useful for samples that are difficult to dissolve or that scatter light, such as powders, gels, and biological tissues [124]. ATR-FTIR has found widespread use in pharmaceutical quality control, environmental monitoring, and the study of biological samples, where it allows researchers to analyze small sample volumes or trace-level contaminants [125, 126].
Furthermore, FTIR spectroscopy plays a crucial role in chemical reaction monitoring. By continuously collecting spectra during the course of a reaction, researchers can track changes in molecular composition and gain insights into reaction kinetics, mechanisms, and intermediate species. This is especially useful in catalysis, where understanding how reactants, products, and catalysts interact at a molecular level is essential for optimizing reaction conditions. Real-time FTIR monitoring has become an indispensable tool in both academic research and industrial applications, helping scientists design more efficient chemical processes [127].
Despite its broad utility, FTIR spectroscopy has some limitations. One of the primary challenges is the interpretation of overlapping spectral peaks, especially in complex mixtures, where different molecular vibrations can occur at similar frequencies. Additionally, the technique is less effective for detecting molecules that lack a permanent dipole moment, as these do not strongly absorb infrared radiation. However, these challenges are often mitigated by combining FTIR with other complementary techniques, such as Raman spectroscopy or NMR spectroscopy, to provide a more complete understanding of the molecular structure.
For that reason, Fourier-transform infrared spectroscopy is an essential analytical technique in modern science, offering deep insights into molecular structure, chemical composition, and interactions. Its versatility, ease of use, and ability to analyze a wide range of sample types have made it a cornerstone in many scientific disciplines, from chemistry and materials science to biochemistry and environmental monitoring. As FTIR technology continues to advance, with improvements in resolution, sensitivity, and data analysis, it will undoubtedly remain a key tool for researchers seeking to understand the molecular world.
2.4.1. Case study of Fourier-transform infrared spectroscopy for cytochrome c in lipid–protein interaction analysis context
A significant example of lipid–protein interaction analysis using FTIR involves studying the interaction between cytochrome c and phospholipid membranes [128]. Cytochrome c is a membrane-associated protein that plays a critical role in the mitochondrial electron transport chain and apoptotic pathways [129]. Its interaction with the inner mitochondrial membrane, particularly with cardiolipin, a specialized phospholipid, is crucial for its biological activity [130]. Using FTIR spectroscopy, researchers aimed to investigate how cytochrome c undergoes conformational changes when interacting with lipid membranes and how these changes impact both protein structure and lipid behaviour.
The exemplified FTIR study involved the interaction of cytochrome c with anionic lipid membranes, including those composed of dimyristoyl, dipalmitoyl, dioleoyl phosphatidylglycerols, and bovine heart cardiolipin. FTIR analysis revealed that the secondary structural changes in cytochrome c upon binding to negatively charged lipid membranes were minimal, as indicated by the Amide I band in the spectrum [131]. There was no significant temperature dependence in the difference spectra between free and bound cytochrome c at temperatures below denaturation. However, in the denatured state, the lipid-bound protein exhibited more disorder compared to the free protein, as indicated by an increased intensity in the random structure region around 1648 cm\(^{-1}\). This suggests that the lipid environment influences the protein's conformational flexibility.
A striking observation was the marked reduction in the protein's denaturation temperature, which dropped by approximately 25–30\(^{\circ}\)C upon lipid binding. This decrease followed a distinct pattern, aligned with the lipid phase transition temperatures, with complexes involving dioleoyl phosphatidylglycerol showing a decrease around 7\(^{\circ}\)C lower than those with dipalmitoyl phosphatidylglycerol. This highlights the profound influence of lipid composition on protein stability.
In addition to these findings, substantial changes were observed in the amide proton exchange dynamics of cytochrome c when monitored in D\(_2\)O. The bound form of the protein exhibited an increase in amide deuteration rates by up to two orders of magnitude across a temperature range of 10 to 40\(^{\circ}\)C, compared to the unbound protein, whose exchange rates remained almost unaffected by temperature. Moreover, the extent of exchange differed between the free and bound states, suggesting that lipid binding alters the protein's exposure and exchange kinetics.
Crucially, a tertiary structural transition was identified in the membrane-bound protein, observed as a sharp change in deuterium exchange rates in Arrhenius plots. This transition occurred at temperatures between 22 and 29\(^{\circ}\)C, varying with the lipid type, and notably well below the protein's denaturation point. The lipid's physical state played a pivotal role in determining the transition temperature, with lipids in the fluid phase showing transitions 7\(^{\circ}\)C lower than those in the gel phase. For dimyristoyl phosphatidylglycerol complexes, the transition temperature was closely linked to the lipid chain-melting transition at 27–28\(^{\circ}\)C.
In summary, cytochrome c's structural stability, both in terms of resistance to denaturation and tertiary structural changes as reflected in amide deuteration rates, is intricately tied to the physical state of the lipid to which it is bound. The control exerted by lipid phase transitions, particularly in dimyristoyl phosphatidylglycerol complexes, underscores a direct coupling between lipid state and protein structure. This coupling mechanism hints at broader regulatory possibilities for membrane-bound enzymes, where the dynamic interplay between lipids and proteins could fine-tune enzymatic activity in response to changes in membrane properties [132].
3. Integration of neutron techniques in lipid–protein interaction studies
Neutron- and photon-based techniques, such as neutron scattering, X-ray diffraction, and fluorescence spectroscopy, offer unique and synergistic tools to probe these interactions with high resolution and specificity [133]. The integration of neutron and photon techniques in lipid–protein interaction studies has advanced our understanding of the physical properties and conformational changes that govern membrane-associated processes, revealing crucial details about both lipids and proteins in their native environments [134,135].
Neutron techniques, particularly SANS and neutron reflectometry, are powerful for investigating the structural organization of lipid–protein systems. Neutrons interact differently with atomic nuclei compared to photons, and their sensitivity to lighter elements like hydrogen makes them particularly useful in studying biological materials. A key advantage of neutron scattering is its ability to distinguish between different isotopes, especially hydrogen and deuterium, enabling contrast variation experiments [136]. In lipid–protein interactions, this contrast variation technique allows researchers to selectively highlight or obscure specific components of the system — lipids or proteins — by using deuterated forms of either molecule. For example, by deuterating the lipid component and keeping the protein hydrogenated, neutron scattering can provide detailed structural information about how proteins embed into or associate with lipid bilayers [137,138].
In studies of integral membrane proteins, SANS and Small-Angle Neutron Diffraction (SAND) open a window into the microscopic world, allow one to explore the intricate architecture of biological membranes and molecular interactions, revealing subtle structural shifts that hold the key to understanding complex biological processes [139], such as in Figure 6, where the primary results of SANS and SAND are exemplified by the bilayer thickness parameters. The shifts in the lipid headgroup positions allow for calculating the diffuse height distribution width (D\(_{HH}\)) as a function of varying cholesterol and melatonin concentrations in 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC). Additionally, the Neutron Scattering Length Density (NSLD) profiles presented in Figure 6b reveal a clear distinction in how cholesterol and melatonin interact with di-monounsaturated DOPC compared to fully saturated DPPC. For DOPC, an increase in bilayer thickness occurs only with the incorporation of 29 mol% cholesterol and 9 mol% melatonin [140].
Photon-based techniques such as X-ray diffraction, fluorescence spectroscopy, and CD spectroscopy complement neutron methods by offering high-resolution structural and dynamic data. X-ray diffraction, in particular, provides atomic-scale resolution of lipid–protein complexes, making it ideal for crystallographic studies of membrane proteins [141,142]. By using X rays,
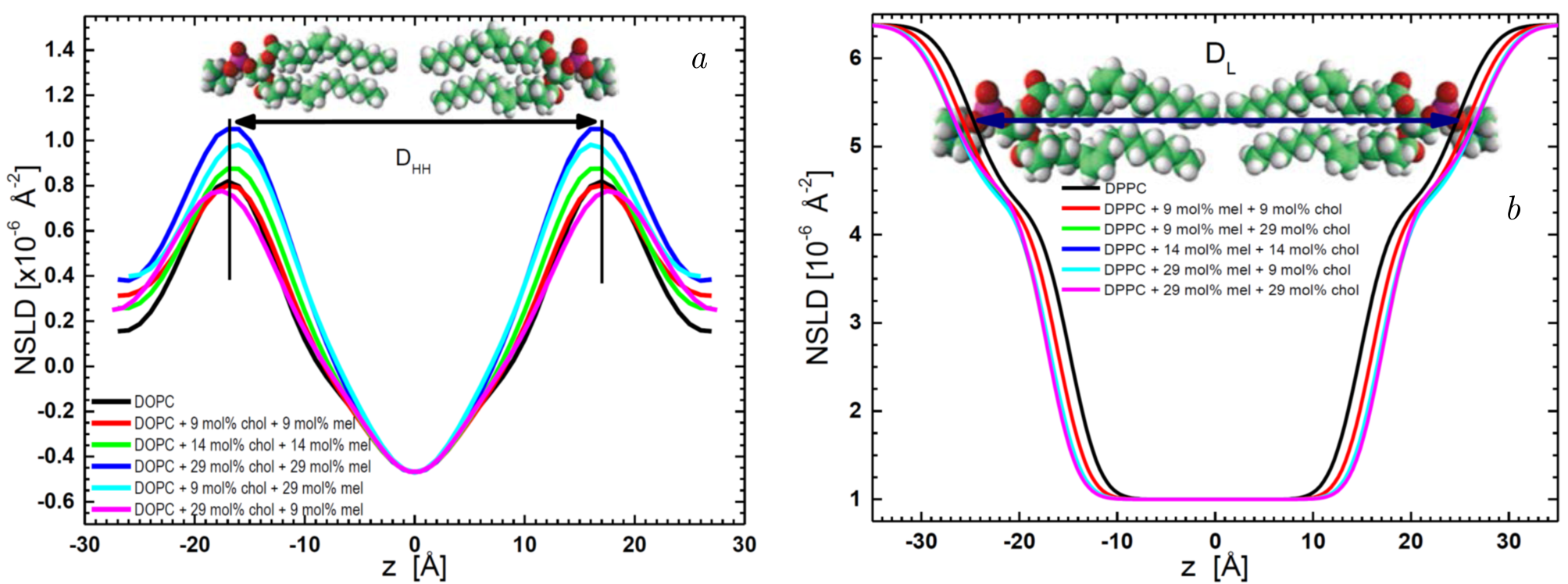
Figure 6. Neutron Scattering Length Density profiles of (\(a\)) DOPC bilayers and (\(b\)) DPPC bilayers with varying compositions of cholesterol and melatonin. Profiles were obtained using 8% D\(_{2}\)O and 100% D\(_{2}\)O hydration, respectively. The bilayer thickness parameter, D\(_{HH}\), represents the distance between the bilayer peaks, while the D\(_L\) thickness corresponds to the lipid headgroup-water interface positions, as indicated by the arrows. Adopted from Kondela et al. [140], the figure is reprinted with the kind permission of the publisher, license number 5935741028308.
researchers can generate diffraction patterns that reveal the arrangement of lipids and proteins in a crystal lattice, offering insights into the interactions that stabilize these structures. One of the challenges of studying membrane proteins with X-ray diffraction, however, is the difficulty in crystallizing them in lipid environments. Nonetheless, the development of lipidic cubic phase crystallization methods has allowed researchers to overcome this challenge, facilitating the structural determination of G protein–coupled receptors, ion channels, and other membrane proteins in native-like lipid bilayers [143].
The integration of neutron and photon techniques provides a more comprehensive understanding of lipid–protein interactions than any single method could achieve alone. For example, while neutron scattering offers insight into the overall architecture of lipid–protein systems, X-ray diffraction provides atomic-level details, and fluorescence spectroscopy reveals dynamic processes in real time. This combination allows researchers to address questions about membrane structure and function from multiple perspectives.
3.1. Neutron scattering and fluorescence spectroscopy in membrane protein insertion studies
Neutron scattering, particularly SANS, and fluorescence-based techniques, such as FRAP, offer powerful tools to investigate membrane protein insertion and diffusion within lipid bilayers [144]. SANS provides structural insights into the positioning and conformation of proteins as they integrate into lipid membranes. This technique is particularly effective due to the contrast variation method, where selective deuteration of lipid or protein components enhances the visibility of specific structures [145]. In parallel, fluorescence-based techniques like FRAP allow researchers to observe the dynamic behavior of proteins as they diffuse laterally within the membrane, revealing important details about protein mobility and lipid–protein interactions [146].
For example, studies on antimicrobial peptides, such as LL-37, used SANS to analyze how these peptides disrupt lipid bilayers [147,148,149], while FRAP revealed how the peptides diffuse within the membrane after insertion [150]. The combination of SANS and fluorescence provides both static structural information and dynamic interaction data, crucial for understanding how such peptides function by permeabilizing membranes. These complementary techniques offer a more comprehensive view of the mechanisms underlying peptide–membrane interactions, as demonstrated in research on antimicrobial peptides in model membranes.
3.2. Small-angle X-ray scattering and small-angle neutron scattering for probing lipid–protein complexes
The combination of Small-Angle X-ray Scattering (SAXS) and SANS has proven invaluable for probing the overall shape, size, and structural organization of lipid–protein complexes. SAXS provides information on the global conformation of these complexes in solution, while SANS, through selective deuteration of lipids or proteins, provides contrast that allows for the differentiation of lipid and protein components within the same system [151].
The possibility of detailed structural information while employing specific deuteration lies in the ability to selectively highlight or obscure certain molecular components within complex systems. This technique enables researchers to isolate structural details, such as lipid bilayer thickness, the spatial organization of proteins within membranes, and protein-induced perturbations in lipid arrangement. For instance, deuterated lipids improve the contrast between lipid and protein regions, facilitating the examination of protein insertion depth, orientation, and conformational changes in lipid bilayers. Furthermore, deuteration permits the analysis of subtle structural differences, such as the distribution of lipid headgroups and tails, as well as interfacial water dynamics. These advances are critical for elucidating the structural principles governing lipid–protein interactions.
In a study reported by Kurakin et al. [152] for the effects of calcium ions on the structural parameters of DPPC membranes incorporating amyloid-beta peptide, SANS and SAXS techniques were employed. The analysis covered a wide range of peptide concentrations (0.01–5 mol% peptide-in-lipid fractions) and calcium ion concentrations (0–10 mM) within the gel phase of DPPC at 20\(^{\circ}\)C. Figure 7 illustrates the SAXS and SANS scattering profiles along with the optimal fits for extruded DPPC (1 wt%) containing varying concentrations of A\(\beta \)(25–35) (Figure 7b and c), and DPPC (1 wt%) samples with A\(\beta \)(25–35) (1 mol%) in the presence of different calcium ion concentrations (Figure 7d and e). Across all peptide and ion concentrations, the scattering curves exhibited typical ULV patterns, with no Bragg peaks observed. As peptide concentration increased, a systematic shift in the scattering minimum towards lower \(q\) values was detected (Figure 7b, arrows), indicating an increase in ULV radii values with higher peptide concentrations in the absence of calcium ions.
3.3. Neutron reflectometry sheds light on membrane structure
A study conducted by Belička et al. [153] employed specular neutron reflectometry to investigate the structure and organization of DPPC lipid bilayers, offering crucial insights into membrane dynamics. They examined a supported lipid bilayer deposited on a solid substrate and a floating bilayer, specifically focusing on how the internal structure changes with temperature, moving from a gel phase at 25\(^{\circ}\)C to a liquid-crystalline phase at 55\(^{\circ}\)C. The scattering data was modeled using a neutron scattering length density profile, revealing that lipid molecules behave differently in supported versus floating bilayers.
The reflectivity profiles showed high structural resemblance between the floating bilayer and unilamellar vesicles, which are commonly used as models for cell membranes. However, the study also uncovered key distinctions, particularly the impact of bilayer undulations and substrate interactions in supported systems. In the liquid-crystalline phase, the flexibility and disorder of the lipids increased, leading to significant changes in bilayer thickness and distribution of lipid components.
Moreover, the study provided detailed information on the separation between the head and tail regions of the lipid molecules, which is crucial for understanding the membrane's role in protein insertion and interaction. The insights gained through neutron reflectometry offered a more refined understanding of lipid bilayer behavior in biomimetic systems, contributing to the broader understanding of membrane dynamics and their functional role in biological processes [153].
4. Quasi-Elastic Neutron Scattering (QENS) as neutron-based tool in studying lipid mobility alterations induced by transmembrane proteins
The study by Ebersberger et al. [154] investigates the influence of transmembrane peptides on lipid dynamics within large unilamellar vesicles composed of DMPC lipids. Using a combination of quasielastic neutron scattering and MD simulations, the researchers explored short-range dynamics on nanosecond time scales and nanometer length scales. The study specifically examined the interaction between the transmembrane sequence of the transferrin receptor (TFRC) protein and the lipids, revealing key insights into lipid mobility and peptide effects on membrane dynamics.
The findings demonstrate that the presence of the TFRC transmembrane peptides significantly restricts the lateral mobility of lipids. This reduction in mobility was quantitatively
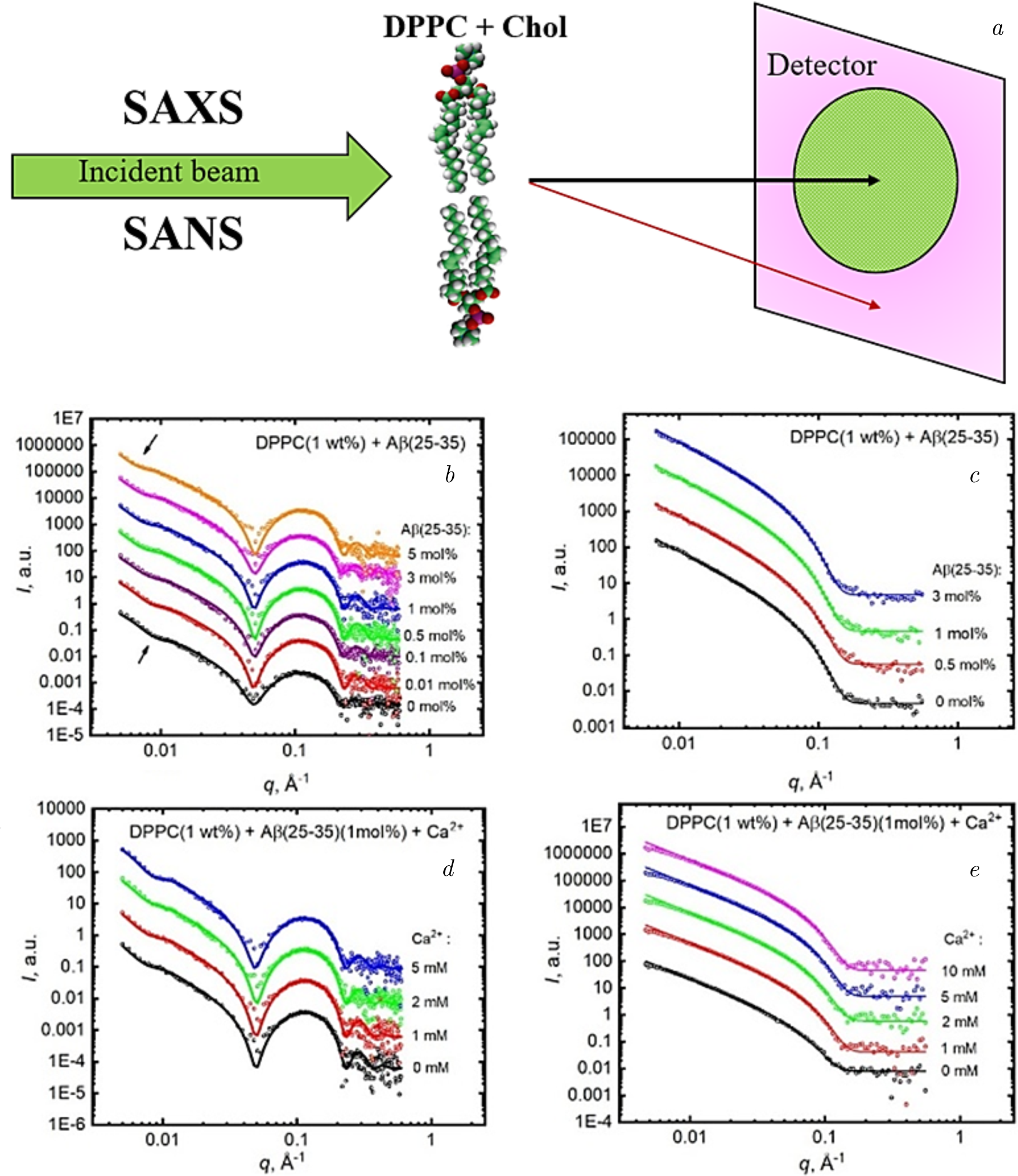
Figure 7. (\(a\)) Schematic representation of the SAXS and SANS processes for DPPC \(+\) A\(\beta \)(25–35) (\(b\)) SAXS and (\(c\)) SANS intensities \(I\) as a function of the scattering vector \(q\) for DPPC (1 wt%) \(+\) A\(\beta \)(25–35) unilamellar vesicles (ULVs) containing various concentrations of A\(\beta \)(25–35): 0, 0.01, 0.1, 0.5, 1, 3, and 5 mol%. (\(d\)) SAXS and (\(e\)) SANS intensities \(I\) for DPPC (1 wt%) \(+\) A\(\beta \)(25–35) (1 mol%) ULVs containing different concentrations of calcium ions: 0, 1, 2, and 5 mM, while the concentration of A\(\beta \)(25–35) remains constant at 1 mol%. Measurements were performed at \(T=20^{\circ}\)C in the gel phase of DPPC. Adapted from Kurakin et al. [152], the figure is reprinted with the kind permission of the publisher, license number 5935740494492.
measured through QENS and validated by MD simulations, emphasizing the complementary nature of these techniques [155]. The apparent self-diffusion coefficient of the peptides in the membrane was also determined, offering a detailed understanding of the interaction dynamics. Notably, the researchers estimated the "radius of influence" of the peptides on lipid long-range dynamics, integrating experimental and simulation data to provide a comprehensive model [156].
This approach highlights the utility of neutron scattering techniques in characterizing lipid–protein interactions at a molecular level. By incorporating contrasting methods (deuterated versus protonated lipids), the study successfully decoupled the dynamics of lipids and peptides, offering an advanced framework for analyzing membrane-associated processes critical for cellular functions such as signaling and molecular transport [157].
These findings provide valuable insights for understanding membrane biophysics, particularly in contexts where protein–lipid interactions play a pivotal role, such as neurodegenerative diseases and drug design. The integration of high-resolution experimental data with computational simulations exemplifies a robust methodology for studying complex biological systems.
5. Challenges and limitations in resolving membrane complexity
Spectroscopic techniques have revolutionized the study of lipid–protein interactions, offering real-time, non-invasive insights into the dynamic properties of biological membranes. However, one of the major challenges in using spectroscopy to study lipid–protein systems is the inherent complexity and heterogeneity of biological membranes. Cellular membranes are highly dynamic, consisting of diverse lipid species and proteins that interact in complex and sometimes transient ways. This heterogeneity is particularly pronounced in systems such as lipid rafts, which are small, transient domains within the membrane enriched in cholesterol and sphingolipids [158]. The spatial and temporal resolution required to study these dynamic microdomains is often beyond the capabilities of conventional spectroscopic techniques [159].
Spectroscopic methods like fluorescence and Raman spectroscopy, while powerful, typically lack the spatial resolution necessary to distinguish between different microdomains within the membrane, especially in live-cell or in vivo studies [160]. Additionally, biological membranes are not uniform, and their complexity is further increased in pathological conditions such as NDDs, where lipid composition and membrane integrity are disrupted [161]. For instance, the formation of amyloid plaques in Alzheimer's disease or Lewy bodies in Parkinson's disease alters the membrane environment, leading to increased heterogeneity that complicates spectroscopic analysis. Resolving these complex systems often requires a combination of multiple techniques or the integration of advanced microscopy with spectroscopy to achieve higher resolution.
Moreover, protein–lipid interactions are often influenced by the curvature and phase behavior of membranes, which can vary significantly across different regions of the cell or even within artificial lipid bilayers used in experimental setups [162]. This presents a challenge when trying to generalize findings from simplified model systems to the complex, dynamic environments of living cells. Spectroscopic techniques must therefore continue to evolve to better resolve membrane heterogeneity and capture the full spectrum of lipid–protein interactions occurring in real time within these complex systems.
5.1. Overcoming fluorescence quenching and photobleaching
Fluorescence-based spectroscopic techniques methods are not without their limitations, with fluorescence quenching and photobleaching being two of the most prominent challenges. Fluorescence quenching occurs when a fluorescent molecule loses its ability to emit light due to interactions with nearby molecules or changes in the environment, leading to a loss of signal. This is particularly problematic in lipid–protein studies, where proteins may insert into or disrupt the membrane, altering the local environment in ways that quench the fluorescence of lipid-bound probes.
Similarly, photobleaching — where prolonged exposure to light causes the fluorescent molecules to degrade and lose their ability to fluoresce — poses a significant limitation for time-resolved studies. Photobleaching not only reduces the overall signal but also limits the duration of experiments, making it difficult to track dynamic interactions over long periods. In the context of NDDs, where protein aggregation and membrane disruption occur over extended time scales, overcoming photobleaching is crucial for accurately capturing the progression of these events.
To mitigate these technical limitations, several strategies have been developed [163]. One approach to reduce fluorescence quenching is the careful selection of fluorescent probes that are less sensitive to environmental changes. In some cases, using longer-wavelength fluorophores, which are less prone to quenching by nearby molecules, can improve signal stability. Additionally, advanced techniques such as fluorescence lifetime imaging microscopy allow researchers to measure the fluorescence decay rates rather than relying solely on intensity, offering a more robust method to detect interactions even in the presence of quenching.
Photobleaching can be minimized by using lower light intensities or applying intermittent illumination rather than continuous exposure, which reduces the cumulative light exposure on the sample [164]. More advanced techniques, such as Total Internal Reflection Fluorescence (TIRF) microscopy or stimulated emission depletion microscopy, also help overcome the limitations of photobleaching by selectively illuminating only the region of interest, thereby preserving fluorescence in the surrounding areas. The use of newer, more photostable fluorophores and the development of novel light sources such as pulsed lasers further improve the longevity and accuracy of fluorescence-based assays [165].
5.2. Quasi-elastic neutron scattering
Quasi-Elastic Neutron Scattering (QENS) offers remarkable insights into lipid dynamics and mobility; however, it encounters limitations that restrict its broader applicability. A key challenge lies in the inherently low signal-to-noise ratio, stemming from the weak scattering intensity of hydrogen-containing samples [166]. This necessitates the use of deuterated lipids, which can be expensive and technically demanding to produce. Additionally, the temporal resolution of QENS may not be sufficient to fully capture ultrafast molecular motions, potentially omitting crucial dynamic events in lipid–protein systems. Data interpretation also presents a significant hurdle, as QENS outputs require complex computational modeling. Such models often simplify biological membranes, introducing uncertainties that may limit the fidelity of the results.
5.3. Neutron reflectometry
Neutron Reflectometry (NR) provides exceptional depth-resolved structural information about membranes, yet it faces practical limitations. The technique requires planar membrane samples, which might not replicate the natural curvature and dynamic properties of biological membranes. The method's low throughput is another limitation; the extensive measurement times and sensitivity to environmental disturbances make it less practical for high-throughput or dynamic studies. Additionally, the reliance on contrast variation via deuteration can limit experimental designs, especially when isotopically labeled components are unavailable or too costly for extensive studies [167].
5.4. Small-angle scattering techniques (SAXS and SANS)
Small-Angle X-ray Scattering (SAXS) and Small-Angle Neutron Scattering (SANS) are invaluable for investigating global structural properties of lipid–protein complexes. However, their limited spatial resolution restricts the detailed exploration of fine molecular arrangements and transient structural dynamics. Furthermore, both techniques require relatively high sample concentrations to achieve adequate scattering intensities, which may not reflect physiological conditions, especially for rare or fragile biological samples. Radiation damage is another concern, particularly in SAXS, where extended exposure to X~rays can alter the sample's structure, complicating data interpretation [168].
5.5. Fluorescence and circular dichroism spectroscopy
Fluorescence spectroscopy and Circular Dichroism (CD) spectroscopy remain pivotal tools for monitoring lipid–protein interactions and conformational changes. Nonetheless, both face limitations. Fluorescence-based techniques are highly sensitive to environmental factors, with quenching and background interference frequently complicating data acquisition. Moreover, these methods generally provide averaged structural information, making it difficult to resolve heterogeneous populations or transient interaction states. In fluorescence spectroscopy, photobleaching — where prolonged illumination degrades fluorescent probes—can significantly limit the duration and quality of experiments. For CD spectroscopy, the spectral overlap between lipids, proteins, and other biomolecules often complicates the interpretation of secondary structural transitions.
5.6. Raman and FTIR spectroscopy
Raman and Fourier Transform InfraRed (FTIR) spectroscopy are complementary techniques that offer molecular-level insights into the chemical composition of lipid–protein systems. Nevertheless, Raman spectroscopy is hindered by weak signal intensity, often requiring the use of high-power lasers that risk heating or damaging the sample. FTIR faces a similar challenge with overlapping spectral peaks, as signals from lipids and proteins can obscure one another, complicating quantitative analyses. Both techniques are also highly sensitive to environmental interference; for example, water vibrations can mask relevant signals, particularly in hydrated membrane systems.
5.7. Integration with computational models
Given the complexity of lipid–protein systems and the limitations of experimental techniques, the integration of spectroscopy with computational models, particularly molecular dynamics simulations, has become increasingly important. MD simulations allow researchers to model lipid–protein interactions at atomic resolution, providing detailed insights into the behavior of these systems over time. Combining experimental spectroscopic data with MD simulations enables a more comprehensive understanding of lipid–protein interactions, as each approach complements the other's strengths [169,170].
MD simulations are particularly valuable for interpreting spectroscopic data that may be difficult to resolve due to membrane heterogeneity or transient interactions. For example, while spectroscopy can provide information on changes in membrane fluidity or protein conformation, MD simulations can help to explain the underlying molecular mechanisms driving these changes. This is especially relevant in the study of protein aggregation, where simulations can model the early stages of oligomer formation and their interactions with lipid bilayers, which are often difficult to capture experimentally [171].
In the case of tau protein interactions with phospholipid bilayers, a relevant topic in Alzheimer's research, the integration of spectroscopic techniques with MD simulations has proven to be particularly fruitful. Time-resolved spectroscopy can track the kinetics of tau aggregation on the membrane surface, while MD simulations can model how specific lipid–protein interactions influence the aggregation pathway [172]. This combination of approaches provides a more detailed picture of the mechanisms by which tau disrupts membrane integrity, contributing to neurodegeneration in AD [173].
Overall, the integration of spectroscopy with computational models enhances our ability to probe lipid–protein systems in ways that would be difficult or impossible using experimental methods alone. By bridging the gap between experimental observation and theoretical modeling, this combined approach allows for a more holistic understanding of the dynamic and complex nature of lipid–protein interactions, particularly in the context of NDDs. Moving forward, continued advancements in both experimental and computational methodologies will be essential for overcoming the remaining challenges in this field and uncovering new therapeutic targets for neurodegenerative diseases.
6. Emerging trends and future directions
As the study of lipid–protein interactions continues to expand, several emerging trends in spectroscopic techniques, therapeutic implications, and the integration of photonic methods with other analytical approaches are shaping the future of this field. These advances hold particular promise for enhancing our understanding of the molecular mechanisms underlying NDDs such as Alzheimer's and Parkinson's, where lipid–protein interactions play a crucial role in disease progression. The development of more sophisticated techniques is essential for overcoming current limitations, enabling researchers to probe these interactions with greater precision and in real time, while also paving the way for novel therapeutic strategies aimed at stabilizing lipid membranes and preventing toxic protein aggregation.
6.1. Advanced photonic techniques
One of the most exciting areas of development in the field of lipid–protein interactions is the integration of advanced photonic techniques, such as single-molecule fluorescence spectroscopy and real-time imaging. Single-molecule fluorescence spectroscopy has revolutionized the study of biological systems by allowing researchers to observe and track individual molecules in complex environments. This technique provides unprecedented spatial and temporal resolution, making it possible to detect transient interactions between lipids and proteins that may otherwise be obscured in ensemble measurements.
In the context of neurodegenerative diseases, single-molecule fluorescence spectroscopy can be used to monitor the behavior of amyloidogenic proteins like A\(\beta \), \(\alpha \)-syn, and tau at the membrane interface. By labeling individual protein molecules with fluorescent probes, researchers can track their diffusion, aggregation, and interaction with specific lipid species in real time. This approach is particularly useful for studying the early stages of protein aggregation, which are critical for understanding how toxic oligomers form and disrupt membrane integrity in diseases such as Alzheimer's and Parkinson's. Moreover, single-molecule techniques can reveal heterogeneity in protein behavior, offering insights into how different conformational states of a protein may interact differently with lipid bilayers.
In addition to single-molecule fluorescence, the development of real-time imaging techniques is opening new avenues for studying dynamic lipid–protein interactions in live cells. Real-time fluorescence imaging, coupled with techniques like TIRF microscopy, allows researchers to visualize the insertion, aggregation, and diffusion of proteins at the cell membrane with high temporal resolution. These techniques have been instrumental in capturing the dynamic processes underlying protein–lipid interactions, such as the oligomerization of A\(\beta \) and its subsequent insertion into lipid rafts, providing a more detailed understanding of the early pathogenic events in Alzheimer's disease.
Conclusion
Spectroscopic techniques have proven particularly powerful in unraveling the complex dynamics of lipid–protein systems. Raman spectroscopy has been employed to track conformational changes in amyloid-beta (A\(\beta )\) peptides, identifying how specific lipid environments facilitate the transition of these peptides into \(\beta \)-sheet-rich structures associated with plaque formation in Alzheimer's disease. These studies have illuminated how lipid composition, particularly the presence of cholesterol and gangliosides in lipid rafts, serves as a catalyst for the aggregation process. Similarly, fluorescence-based techniques such as FRET have provided detailed spatial and temporal resolution of interactions between proteins and lipids, demonstrating the binding and insertion mechanisms of \(\alpha \)-synuclein into membranes — a critical step in Parkinson's disease pathogenesis. The ability of these techniques to capture dynamic, real-time interactions under physiologically relevant conditions has greatly enhanced our understanding of how specific lipid microdomains act as nucleation sites for pathological protein aggregation.
Fluorescence quenching and Raman studies have revealed that lipid raft disruption, such as through cholesterol depletion, can mitigate aggregation and restore membrane fluidity, offering a potential therapeutic avenue. Furthermore, techniques like circular dichroism and infrared spectroscopy have highlighted how changes in membrane curvature and fluidity induced by protein–lipid interactions influence the kinetics of protein aggregation and oligomerization. These findings provide a comprehensive understanding of how membrane composition directly impacts the structural transitions that underlie disease states.
Complementing these spectroscopic insights, neutron scattering techniques have emerged as indispensable tools for investigating the nanoscale structural and dynamic properties of lipid–protein systems. SANS has enabled high-resolution characterization of lipid bilayers and their interaction with proteins. By employing isotopic contrast variation — leveraging the differential scattering properties of hydrogen and deuterium — SANS provides unique structural information that is unattainable with other techniques. For instance, studies using SANS have revealed how cholesterol modulates bilayer thickness and fluidity, providing insights into its role as a key regulator of protein–membrane interactions. Similarly, neutron reflectometry has been instrumental in mapping the organization of supported lipid bilayers, uncovering subtle temperature-dependent structural changes and their implications for protein binding.
Dynamic investigations of lipid–protein systems have been greatly advanced by QENS, which offers unparalleled insights into the mobility of lipids and their modulation by embedded proteins. By quantifying the radius of influence exerted by transmembrane peptides on lipid dynamics, QENS studies have provided detailed views of the localized effects of peptide insertion on bilayer behavior. MD simulations have further complemented these experimental findings, allowing researchers to simulate lipid–protein interactions at atomic resolution and under varying environmental conditions. Together, these approaches have elucidated how alterations in lipid composition, such as changes in cholesterol or unsaturated lipid content, affect membrane properties, including bending rigidity and lateral diffusion, which are critical for biological function and stability.
The integration of spectroscopic and neutron-based techniques has facilitated a multidimensional approach to lipid–protein research. For example, combining SANS with FRAP has enabled simultaneous exploration of structural and dynamic aspects of lipid–protein systems. This combined methodology has proven particularly valuable in studying antimicrobial peptides, revealing both the structural deformation of membranes and the dynamic recovery processes that follow peptide insertion. Such synergistic approaches not only provide a holistic understanding of lipid–protein interactions but also underscore their potential for guiding the development of therapeutic interventions.
Looking ahead, the convergence of advanced experimental techniques with computational modeling holds significant promise for the field. Molecular dynamics simulations have already demonstrated their potential to unravel the complexities of lipid–protein interactions, offering atomistic insights that bridge the gap between experimental data and theoretical predictions. Future advancements in single-molecule spectroscopy and enhanced neutron instrumentation are poised to provide real-time, high-resolution observations of these interactions under near-physiological conditions. The development of next-generation isotopic labeling techniques and hybrid experimental-computational workflows will further enhance our ability to probe the nuanced interplay between lipids and proteins.
From a therapeutic perspective, these advances have profound implications. By elucidating the structural and functional determinants of lipid–protein interactions, researchers can identify potential drug targets aimed at stabilizing membranes and preventing protein misfolding and aggregation. Lipid-targeting therapies, such as cholesterol-lowering agents or small molecules that disrupt lipid raft integrity, hold significant promise in mitigating the progression of neurodegenerative diseases. Furthermore, the design of anti-aggregation molecules that selectively bind to pathological protein species without disrupting normal cellular function represents a transformative strategy for combating NDDs.
In conclusion, the study of lipid–protein interactions through the lens of spectroscopic and neutron-based methodologies has unveiled critical insights into the molecular mechanisms underlying neurodegenerative diseases. By bridging the structural, dynamic, and functional aspects of these interactions, researchers have laid the groundwork for innovative therapeutic strategies. As experimental technologies continue to evolve and integrate with computational approaches, the field stands poised to make groundbreaking contributions to our understanding of membrane biology and its role in health and disease. These efforts promise to not only advance basic scientific knowledge but also drive the development of novel therapies for some of the most challenging conditions in modern medicine.
Acknowledgements
The authors of the article welcome the establishment of a new multidisciplinary scientific journal NSR at JINR with open access. This journal facilitates the publication of both review and original scientific papers, encompassing a broad spectrum of fundamental and practical research conducted within the JINR Member States and worldwide.
Author contributions
G. M. Arzumanyan: Conceptualization and supervision. Each author participated in the conception and design of the work, as well as drafting and revising the manuscript.
Funding
This work was supported by the JINR theme project “Nanobiophotonics” (# 1147-1).
Conflicts of Interest
The authors declare no conflict of interest.